
Chemical Physics
Machine Learning Interatomic Potentials (MLIPs)
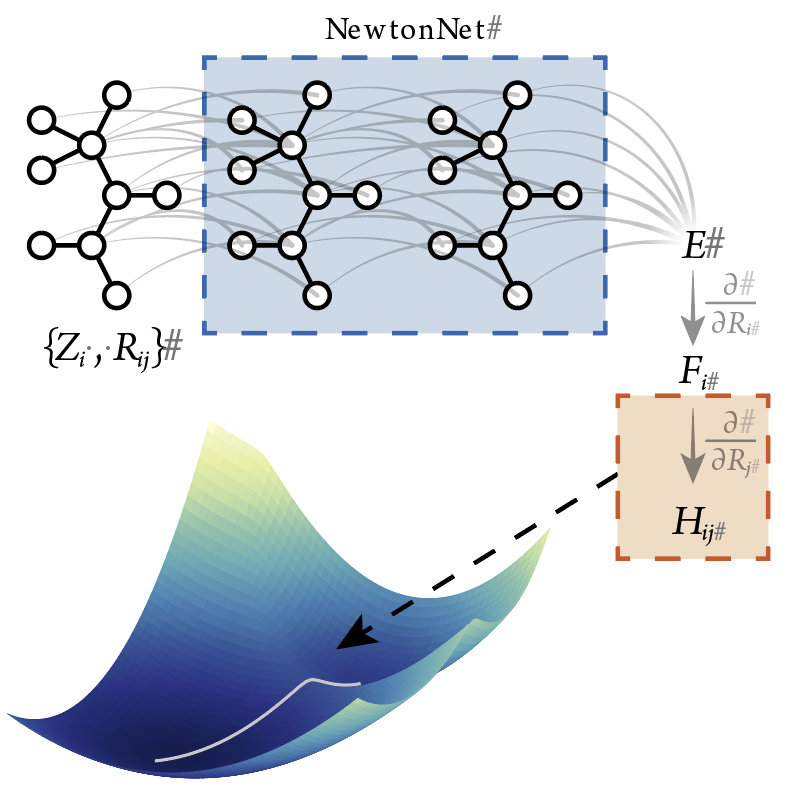 In our group, we develop foundational machine learning interatomic potentials (MLIPs) that go beyond single-point energy predictions to enable more accurate and transferable molecular simulations. A key focus is designing advanced training strategies that enhance MLIP accuracy across diverse chemical environments while maintaining computational efficiency. We explore innovative approaches to extend MLIPs for molecular dynamics (MD) simulations, capturing long-timescale structural and dynamic properties with quantum-level accuracy. Additionally, we integrate MLIPs into transition state pathway predictions, enabling more efficient exploration of reaction mechanisms and energy landscapes. By improving both training methodologies and application strategies, our work expands the capabilities of MLIPs, making them more reliable for complex simulations in chemistry and materials science.
In our group, we develop foundational machine learning interatomic potentials (MLIPs) that go beyond single-point energy predictions to enable more accurate and transferable molecular simulations. A key focus is designing advanced training strategies that enhance MLIP accuracy across diverse chemical environments while maintaining computational efficiency. We explore innovative approaches to extend MLIPs for molecular dynamics (MD) simulations, capturing long-timescale structural and dynamic properties with quantum-level accuracy. Additionally, we integrate MLIPs into transition state pathway predictions, enabling more efficient exploration of reaction mechanisms and energy landscapes. By improving both training methodologies and application strategies, our work expands the capabilities of MLIPs, making them more reliable for complex simulations in chemistry and materials science.
Microdroplet Chemistry

A wide variety of reactions are reported to be dramatically accelerated in aqueous microdroplets, making them a promising platform for environmentally clean chemical synthesis. However, to fully utilize the microdroplets for accelerating chemical reactions requires a fundamental understanding of how microdroplet chemistry differs from that of a homogeneous phase. Our group focuses on developing theory models to explain the observed experimental phenomena by utilizing molecular dynamics simulations to study various system properties. By bridging experimental observations with theoretical insights, we strive to provide a predictive framework for microdroplet chemistry that can guide future experimental designs and applications.
Oil-Water Interface

Mesoscale water-hydrophobic interfaces play a crucial role in various disciplines, yet their molecular properties have remained challenging to characterize due to experimental limitations and conflicting theoretical interpretations. In our group, we develop theory models to explained the experimental observations from a novel interface-selective Raman spectroscopy approach at the oil-water interface. With these developed models, we are able to characterize unique structural and electrostatic properties of mesoscale hydrophobic interfaces, distinguishing them from molecular-scale solvation environments and underscoring their importance in interfacial chemistry and reactivity.
Force Field Development
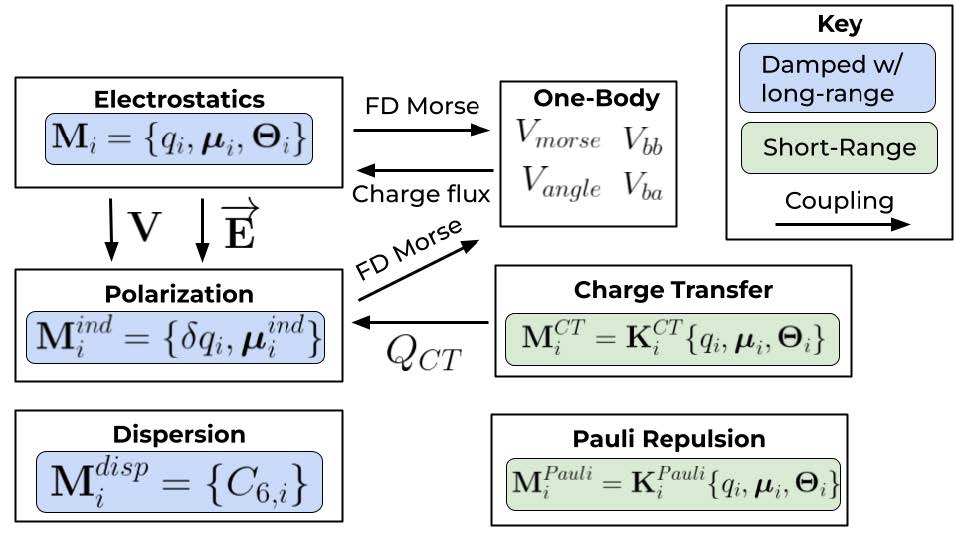
Force fields (FFs) are essential for accurately modeling molecular interactions, and ongoing developments focus on improving their transferability, accuracy, and chemical insight. In our group, we are working on designing more generalized FFs that aim to achieve the accuracy of different levels of theory while maintaining efficiency. One direction focuses on improving intramolecular energy terms, as seen in the development of 1B-UCB, a simple yet accurate one-body energy and dipole moment surface model that integrates seamlessly into automated workflows and extends beyond water to other small molecules. Another effort refines intermolecular interactions through the Completely Multipolar Model (CMM), which incorporates environment-dependent atomic polarizability to better capture polarization and charge transfer effects in ion–water and ion–ion interactions. Additionally, we are advancing methods for identifying bond dissociation points by introducing the Wiberg–Mayer bond flux, a more direct metric for bond breaking that provides greater chemical insight into reaction mechanisms. Together, these efforts enhance FF accuracy and generality by addressing intramolecular energy, complex multipolar interactions, and fundamental bond reactivity.
M-Chem: An MD Engine

The main goal of M-Chem is to perform classical MD simulations and further extend to ab-initio MD and hybrid quantum mechanics/molecular mechanics functionalities. In the future, M-Chem will be used to perform simulations on various biomolecular systems ranging from thousands of atoms to over several hundred thousand atoms.

Here at THG lab, we operate at the intersection of computational chemistry, biophysics, and machine learning. Our main interests are around the development and application of novel methodologies to solve critical challenges in protein modeling and drug discovery. A central theme of our research is the synergistic integration of rigorous physics-based models with advanced machine learning techniques. Along this line, we have developed unified software platforms for drug discovery aiming to create and integrate more generalizable and accurate tools than either physics or AI alone could achieve. Currently, our active research topics include the use of Large Language Models (LLMs) for generative chemistry, the curation of high-quality protein-ligand datasets, and the ensemble modeling for Intrinsically Disordered Proteins (IDPs).