Computational Optimization of Electric Fields for Better Catalysis Design
Although the ubiquitous role that long-ranged electric fields play in catalysis has been recognized, it is seldom used as a primary design parameter in the discovery of new catalytic materials. Our group is developing advanced models that permit accurate calculation of electric fields that we have used to computationally optimize biocatalytic performance of synthetic enzymes and supramolecular capsules. We are using electric fields as a unifying descriptor for catalytic design across a range of homogeneous and heterogeneous catalysts. While focusing on electrostatic environmental effects may open new routes toward the rational optimization of efficient catalysts, much more predictive capacity is required of theoretical methods to have a transformative impact in their computational design – and thus experimental relevance – when using electric field alignments in the reactive centers of complex catalytic systems, and defines an ongoing area of research in the Head-Gordon Lab.
Interfaces to Control Chemical Reactivity of Confined Liquids
The ability to control for a chemical reaction process is a fundamental problem that broadly impacts basic energy science efforts in successful creation of next generation fuel cells, photovoltaics, biofuels, and light emitting device technologies. Catalytic acceleration in confined aqueous environments has been found in so called “nano-vessels”, in which organometallic reactions are accelerated when encapsulated within the cavities of water-soluble metal clusters. Biomolecular association rates of proteins confined in the aqueous crowded cell or encapsulated enzymes are found to be catalytically enhanced, and zeolite catalysis succeeds in part by the confined geometry of the active site. Inspired by these examples, our group develops and apply simulation techniques for executing and or accelerating chemical reactions by controlling the altered solvent properties between confining mesoscale interfaces under both equilibrium and dissipative conditions.
Accurate Potential Energy Surfaces
Molecular simulations using classical, molecular mechanic potential energy functions have enjoyed a 50-year history of development, and much has been learned regarding the essential physics that must be captured in order to reproduce observables across a wide range of systems in the condensed phase. While pairwise-additive force fields using fixed, point charge-based electrostatics have dominated this history owing to computational cost, their limitations in transferability are being recognized, owing particularly to the lack of many-body effects, as well as an inherent difficulty in capturing quantum mechanical effects that become important at short intermolecular separation. Our group is advancing new developments for more advanced treatment of molecular interactions manifested as force fields. This work requires the development of novel functional forms that utilize energy decomposition schemes, machine learning-based models, and algorithms that reduce the computational cost, permitting them to become competitive with pairwise-additive models as the standby of condensed-phase simulation.
Intrinsically Disordered Proteins
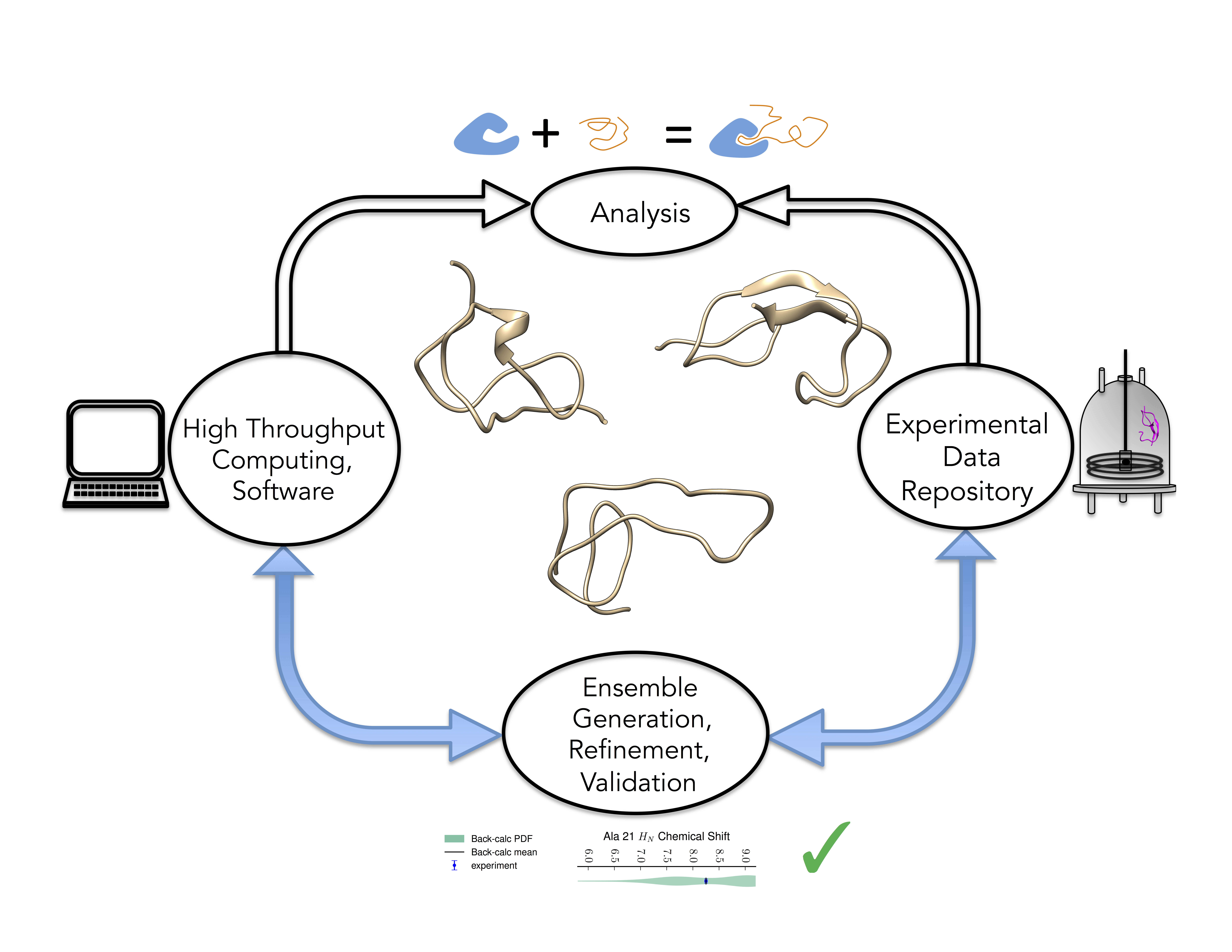 The traditional structure-function paradigm has provided significant insights for well-folded proteins in which structures can be easily and rapidly revealed by X-ray crystallography beamlines. However approximately one third of the human proteome are comprised of intrinsically disordered proteins and regions (IDPs/IDRs) that do not adopt a dominant well-folded structure, and therefore remain “unseen” by traditional structural biology methods. The challenges raised by the “Dark Proteome”, in which determining the diverse conformational substates of IDPs in their free states, in encounter complexes of bound states, and in complexes retaining significant disorder, requires an unprecedented level of integration of multiple and complementary solution-based experiments that are analyzed with state-of-the art molecular simulation, Bayesian probabilistic models, and high throughput computation. Our group envisions how these diverse experimental and computational tools can work together through formation of a “computational beamline” that will allow key functional features to be identified in IDP structural ensembles.
The traditional structure-function paradigm has provided significant insights for well-folded proteins in which structures can be easily and rapidly revealed by X-ray crystallography beamlines. However approximately one third of the human proteome are comprised of intrinsically disordered proteins and regions (IDPs/IDRs) that do not adopt a dominant well-folded structure, and therefore remain “unseen” by traditional structural biology methods. The challenges raised by the “Dark Proteome”, in which determining the diverse conformational substates of IDPs in their free states, in encounter complexes of bound states, and in complexes retaining significant disorder, requires an unprecedented level of integration of multiple and complementary solution-based experiments that are analyzed with state-of-the art molecular simulation, Bayesian probabilistic models, and high throughput computation. Our group envisions how these diverse experimental and computational tools can work together through formation of a “computational beamline” that will allow key functional features to be identified in IDP structural ensembles.
Modeling Electrostatic Phenomena
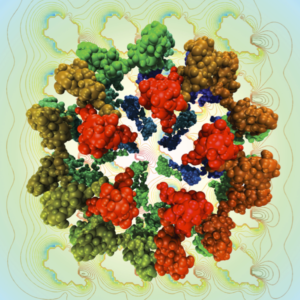
Electrostatic interactions play an important role in molecular simulations. The Poisson-Boltzmann equation (PBE), which describes the charge density using mean-field approximations, can give very accurate results for the electrostatic potential of various systems. Our group has developed PB-SAM, a semi-analytical method for solving a linearized PBE based on ideas from the fast multipole method (FMM) and boundary element (BE) method. Compared with other PBE solvers such as finite-difference (FD) and finite-element (FE), PB-SAM can handle molecules with arbitrary shapes, realize better accuracy, has flexible memory management and a significantly reduced computational cost. We are currently using PB-SAM combined with Brownian dynamics (BD) simulations to study protein-protein interactions and electrocatalytic models of metals with electrolyte.